
Fluorescence is the emission (after an initial photon absorption process) of radiation to a lower state of energy from an excited energy state with the same multiplicity. In here, returning to the lower state is a spin allowed process and happens rapidly by emission of a photon (huF in the above Jablonski diagram). Accordingly, fluorescence occurs about 10-5-10-8 s after the photon absorption. In phosphorescence, the electron spin changes, which is a spin forbidden process. It results in a longer lifetime of the excited state and typically phosphorescence occurs seconds to minutes after the photon absorption (hup in the above Jablonski diagram).
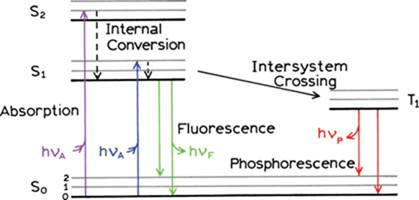
One form of a Jablonski diagram (adapted from J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 3rd Ed., Springer, 2006)
Typically, the fluorescence happens after a series of processes. Initial absorption of a photon leads to a higher vibrational level of an excited state and the molecule rapidly loses its excess of vibrational energy by collision and falls to the lowest vibrational level of that excited state. Thereafter, the molecules lose energy until they reach the lowest vibrational level of the first excited state (S1). From this level, the molecule can return to any of the vibrational levels of the ground state, resulting in the emission of energy in the form of fluorescence. If this process takes place for all the molecules that absorbed light, then the quantum efficiency (no. of photons emitted/ no. of photons absorbed) will be a maximum, unity. However, involvement of other routes such as radiationless transitions lead the quantum efficiency to be less than one.
The absorption and emission processes occur between the lowest vibrational levels of S0 and S1 electronic states, have the same energy gap. Therefore, one can except the overlapping of the emission spectrum to the absorption spectrum for this 0-0 transition. However, in practice due to some interactions with the solvent molecules the absorbing molecules lose some of their energy. As a consequence, the exact overlapping of absorption spectrum to the emission (for 0-0 transitions) is almost impossible. The shift of emission spectrum from that of absorption at 0-0 transition is a key characteristic of fluorescence and it is called “Stokes shift” after the first observation of longer wavelength emission by Sir G.G. Stokes. Another feature of fluorescence is that the same fluorescence emission spectrum is generally observed irrespective of the excitation wavelength. This is as a result of photon emission happening only from the lowest excited state of a given multiplicity as stated from the Kasha’s rule. In addition, the emission spectrum is typically a mirror image of the absorption spectrum. This is due to the fact that the spacing of the vibrational energy levels of the excited states is similar to that of the ground state. As a result of that, the vibrational structures seen in the absorption and the emission spectra are similar, resulting a mirror image.
All fluorescence instruments contain three basic items: a light source, a sample holder and a detector (scheme below). In addition, the wavelength of incident radiation needs to be selectable and the detector signal needs precise manipulation and presentation. For those purposes excitation and emission monochromators are used. Commonly used light sources in fluorescence spectrometry have spectral outputs either as a continuum of energy over a wide range (Ex: Xenon arc, tungsten-halogen lamp) or as a series of discrete lines (ex: mercury lamp). The light passes into the sample cell causes fluorescent emission and the light emitted enters the emission monochromator, which is positioned at a 900 angle from the excitation light path to eliminate background signal and minimize noise due to stray light. Typically, in fluorescence instruments, photomultiplier tubes are used as detectors.
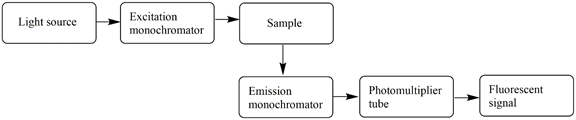 A schematic representation of a fluorimeter
A schematic representation of a fluorimeter
The IC-FAS is equipped with the Thermo Scientific Lumina Fluorescence Spectrometer for steady-state fluorescence measurements. It has the following main specifications.
| Light Source | 150 W Ozone Free Xenon Lamp |
| Detector | R-928 Photomultiplier tube
Silicon photodiode for reference |
| Wavelength Range | 190–1100 nm |
| Accuracy | ±0.5 nm |
| Repeatability | ±0.2 nm |
| Scan Speed | 1–6,000 nm/min |
| Excitation and Emission Spectral Bandwidths | 0.5, 1.0, 2.5, 5.0, 10, & 20 nm |
| Minimum Sample Volume | 0.50 mL (standard 10 mm cuvette) |
The instrument is also supported with Luminous software. At present, we have the capability of analyzing both liquid and solid samples. With the existing solid sampling accessory, the fluorescence intensity can be measured on wide range of solid samples such as thin films, powder and granular samples. For the liquid sample measurements, regular single cell holder or multi-position cell holder can be used depending on the requirement. Especially for the temperature controlled experiments multi-position cell holder can be used for great temperature accuracy and reproducibility.
Sample Preparation Guide
Although fluorescence is a very sensitive technique it can be commonly interfered by trace levels of organic chemicals. For an example, the oils secreted by the skin of the researcher is a possible source of this type of contamination. Therefore, an extreme care should be taken during the sample preparation time. Solvents should be of the highest-level purity obtainable commercially. Suspended particulates such as dust and fibers should not be there in your sample as they will float inside the sampling area of the cuvette leading to scattering effects. In addition, factors like the temperature, pH and light can affect the quality of the result. So, the researcher must take precautions to control these parameters. A minimum of ~0.5 ml sample can be used with an appropriate solvent in a standard 10 mm quartz cuvette for the analysis.
Lecturer-In-Charge
Dr. Janitha Walpita
Click following link for the reservation of the Fluorimeter